
For pharmaceutical product owners, to enhance their patients’ lives is their ultimate goal. Successful technology transfers ensuring product performance and safety before the product goes to market is paramount. For their manufacturing partners – pharmaceutical contract manufacturers – successful technology transfers between clients and ourselves is our ultimate goal. If technology transfers are successful, then product performance in confirmed, supply hiccups are minimized. Technology transfer projects aim for transferring the manufacturing, analytics and packaging, including relevant documentation, knowledge and professional expertise involved in the processes. Quality and safety must remain consistent. Contract manufacturing organizations aim for successful technology transfers while remaining consistent with the safety and quality of their clients’ products. In some cases, technology transfers may not always proceed smoothly, especially for the modified-release oral solid dose (OSD) drugs. Modified-release drug products are designed to release drugs in a controlled manner to achieve desired efficacy and safety profiles. Such modifications have 5 primary objectives, such as:
- Maintaining therapeutic activity for an extended time

- Reducing toxic effects
- Protecting the active substance against degradation due to low pH
- Targeting the active substance to a predefined segment of the gastrointestinal tract for local treatment
- Targeting active substance release at specified time-points
What are the different types of modified or controlled solid oral dose release drugs?
- Sustained-release: where the drug is released slowly at a rate governed by the delivery system
- Controlled-release: where the drug is released at a constant rate and plasma concentrations after administration do not vary with time
- Delayed-release: where the drug is released at a time other than immediately after administration, (i.e. the site of release is controlled)
- Combinations of IR and ER components: where these dosage forms may be designed to deliver drugs in a controlled and predictable manner over a defined period of time or at a predetermined position in the gastrointestinal (GI) tract
Modified-release dosage forms enhance the safety, efficacy, reliability, and convenience of drug therapy by controlling the rate and duration of drug release to control drug actions and to reduce the frequency of drug administration, thus encouraging patient compliance.
What Types of Modified-Release Technologies are Available For Your Product?
There are several known developed technologies for modified-release drugs, such as:
- hydrophilic matrix tablets
- osmotic systems
- coated tablets
- laser-drilled osmotic tablets
- matrix systems
Wurster coating is one of the most recognized methods for developing effective controlled-release formulations. The technology involves bottom-spray fluid bed coating of particles with appropriate actives, excipients, or polymers to modify the drug release. Compared to alternative approaches, Wurster coating is a preferred technology as it enables a highly consistent coating of powders, granules, pellets, or micro-tablets. The accuracy and reproducibility of the coating process result in superior product performance of the controlled-release dose form.
What is Important in Ensuring a Smooth Technology Transfer for Modified-Release OSD Drugs?
Performing technology transfers for modified-release products may get more complex than immediate release dosage form products. Modified-release dosage forms are often designed to maximize a therapeutic effect or minimize side effects to achieve the Quality Target Product Profile (QTPP). It is important to apply the Quality-by-Design (QbD) approach for the development of a modified release product. Under the QbD approach, the critical material attributes and the ranges of critical process parameters for release-controlling drugs should be identified.
The QbD approach promises to build quality into a final product by prospectively designing formulations and processes to meet the predefined QTPP. To design a quality product, this approach calls for identification and control of critical raw material attributes, process parameters, and sources of variability. In the meantime, raw materials and manufacturing processes should be controlled to produce consistent quality in the whole drug life cycle. Tools are available for facilitating the implementation of the QbD, which may include:
- Design of experiments
- Risk assessment
- Process Analytical Technology (PAT)
The design space should also be established to ensure the CQA can meet predetermined specifications during technology transfer and scale-up activities.
When you plan to transfer modified-release products, the product development work can be done at your site or with your CDMO partner. If you have completed the development work, please ensure that information is shared with CDMO partners transparently. By doing so, your CDMO partner can rely on your process knowledge and establish a Normal Operating Range (NOR) during technology transfer or scale-up activities. If the product is still under development, we suggest working with an experienced CDMO partner with plenty of experience in handling modified-release products. The experienced team will handle the products appropriately and make sure the manufacturing process parameters are properly established, and the product quality is in a state of control.
Mini Case Study: Overcoming Poor Bioavailability for Complex Extended-Release Capsule Product
Challenge
A few years ago, Bora developed an extended-release capsule drug (PRODUCT A) with poor bioavailability. The key element of a successful PRODUCT A formulation was to ensure clinically relevant amounts of API will be absorbed from the gastrointestinal tract over a prolonged duration. As a result, a multi-particulate formulation strategy was adopted for the development of the drug. The main advantage of multi-particulate systems is attributed to the consistent gastrointestinal transit characteristics.
PRODUCT A was a multi-particulate formulation designed with an 8-hour extended drug release profile. The formulation contains different formulation components, each exhibiting a unique dissolution profile. In the capsules, several components are filled in with immediate-release (IR) components and extended-release (ER) components.
Methodology
Quality target product profile (QTPP) and the critical quality attributes (CQA) were identified first. The formulation of each component was developed with a relative bioavailability study according to the in vitro dissolution profiles and in vivo performance. The core beads of extended-release components were formulated based on extrusion-spheronization granulation technology. Two layers of coating were applied to the core beads. The first inner layer was a controlled release film, which provided a sustained release of API. The second outer layer was an enteric coat which delayed the release of API in the stomach.
A preliminary risk assessment was conducted for formulation ingredients. The potential critical quality attributes (CQA) for the components included assay, dissolution, and particle size. The particle size was considered an intermediate CQA (iCQA) since it may affect coating and dissolution of API. The risks ranked medium and above require investigations.
Once the formula was confirmed, the process development of each component was initiated. For extended-release components, wet granulation, extrusion-spheronization, drying, sieving, and coating were applied. To gain process understanding early in the development program, the QbD approach was applied to establish both critical process parameters (CPPs) and key parameters, which affected process stability and reproducibility.
Results
Small scale process development for components were focused on each unit operation. Process parameters examined were those identified as high or medium risk in the initial risk analysis. The study design was a four-factor, two-level factorial DOE (16 runs) with the parameters. The design space was explored for each CPP.
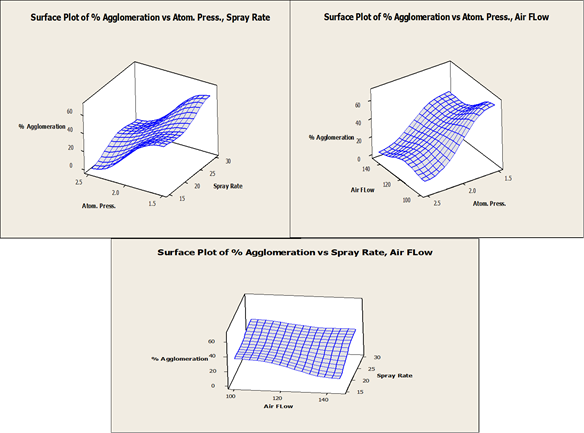
Different atom air and spray rates were studied and the surface plots of % agglomeration were analyzed. The CPP was identified and the particle size of core beads can be well controlled within the established NOR to make sure the product quality is consistent.
Conclusion
The final risk assessment was performed to review the risk level according to study results. The control strategy for components was defined and the acceptance criteria for components were established. Thorough characterization yielded critical process adjustment, which enabled a successful PPQ campaign. Insights gained during process characterization were also utilized for post-launch development.
The choice of manufacturing partner may be one of the more important factors in ensuring a project efficiently and cost-effectively progresses to commercialization. When your partner has significant experience with late-stage work – and a diverse toolkit for accommodating extended release drugs – they can better anticipate and avoid issues that could be problematic for technology transfer or scale-up.
Ultimately, the technology transfer of extended release drugs is filled with risk and has many potential areas for issues to surface; your CDMO partner should not be among these challenges.
Bora has an experienced team to handle complex manufacturing processes for your extended release drugs, including organic solvent solution handling; Wurster coating system, extruder, and spheronizer. Those are standard processes when you are considering an efficient, flexible and fast process for forming uniform modified released particles.



